sMolBoxes: Dataflow Model for Molecular Dynamics Exploration
Pavol Ulbrich, Manuela Waldner, Katarína Furmanová, Sérgio M. Marques, David Bednář, Barbora Kozlikova, Jan Byška
View presentation:2022-10-19T21:21:00ZGMT-0600Change your timezone on the schedule page
2022-10-19T21:21:00Z
Prerecorded Talk
The live footage of the talk, including the Q&A, can be viewed on the session page, DNA/Genome and Molecular Data/Vis.
Fast forward
Abstract
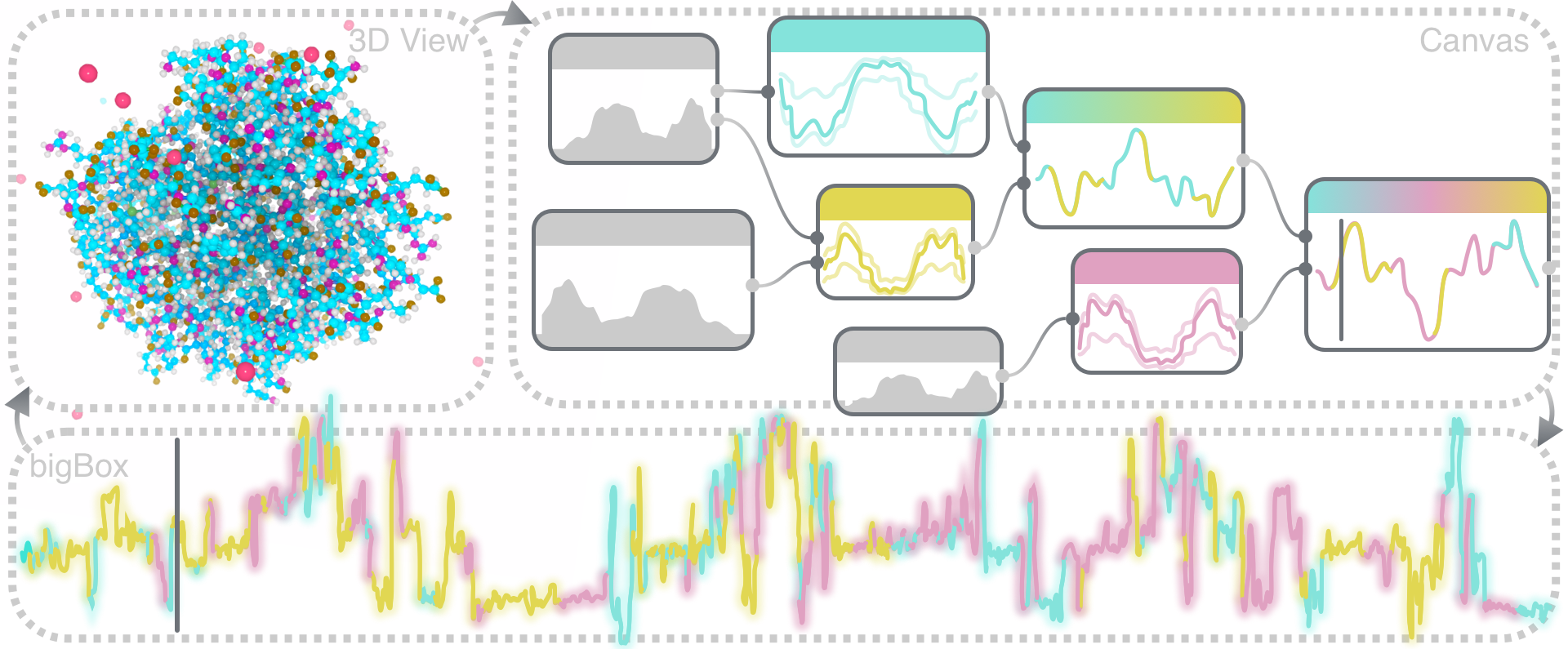
We present sMolBoxes, a dataflow representation for the exploration and analysis of long molecular dynamics (MD) simulations. When MD simulations reach millions of snapshots, a frame-by-frame observation is not feasible anymore. Thus, biochemists rely to a large extent only on quantitative analysis of geometric and physico-chemical properties. However, the usage of abstract methods to study inherently spatial data hinders the exploration and poses a considerable workload. sMolBoxes link quantitative analysis of a user-defined set of properties with interactive 3D visualizations. They enable visual explanations of molecular behaviors, which lead to an efficient discovery of biochemically significant parts of the MD simulation. sMolBoxes follow a node-based model for flexible definition, combination, and immediate evaluation of properties to be investigated. Progressive analytics enable fluid switching between multiple properties, which facilitates hypothesis generation. Each sMolBox provides quick insight to an observed property or function, available in more detail in the bigBox View. The case studies illustrate that even with relatively few sMolBoxes, it is possible to express complex analytical tasks, and their use in exploratory analysis is perceived as more efficient than traditional scripting-based methods.